
The Office of Pharmaceutical Quality (OPQ) within FDA’s Center for Drug Evaluation and Research (CDER) has published its fiscal year 2020 report on the state of pharmaceutical quality.
Read the full report (PDF) on fda.gov here.
The report offers a look into various quality indicators and trends that are of particular interest given a year of enormous challenges for the FDA. The FY2020 report offers a pertinent analysis of the impact the COVID-19 pandemic has had across the pharmaceutical industry—specifically on its supply chains and the quality of its products.
Among other areas of quality, the report provides information on inspectional classification outcomes, product performance, product testing results, and recalls. FDA uses the State of Pharmaceutical Quality, in part, to inform regulatory decision-making and surveillance activities.
Here, we distill some of the most significant findings contained in the report.
1. The total number of drug manufacturing sites in CDER’s Site Catalog decreased by 1.2%.
Excluding medical gas manufacturers and newly registered hand sanitizer manufacturers, the FY2020 CDER Site Catalog has 4,221 drug manufacturing sites, a small decrease (-1.2%) from 4,273 in FY2019.
- The top five countries by the number of manufacturing sites in the FY2020 CDER Site Catalog (United States, India, China, Germany, and Canada) all saw a net decrease in the number of sites.
- "The South Korean site inventory was most dynamic (loss of 34% and gain of 32%), indicating its status as an emerging source of pharmaceutical products."
- At the time of the report’s publishing, there were 1,623 new registrants from around the globe considered “manufacturers” in the FY2021 CDER Site Catalog, 94% of which are hand sanitizer manufacturers.
2. FDA used Mutual Recognition Agreements (MRAs) and its authority to obtain records for sites in advance or in lieu of inspections due to the pandemic.
In 2012, the Food and Drug Administration Safety and Innovation Act gave FDA new authorities under the Food, Drug, and Cosmetic Act §704(a)(4) to request records or other information from firms in advance of or in lieu of an inspection.
(Read FDA’s Staff Manual for requesting records pursuant to this authority here.)
- “FDA surveillance history, requests for records, and inspection reports obtained through the MRAs were all used to mitigate risk and enable regulatory actions.”
- “For submissions that were deemed mission-critical, some inspections were still performed under difficult COVID-19 travel restrictions.”
- “As a result of utilizing all of these approaches, CDER completed facility assessments to meet User Fee dates over 90% of the time and reduced the need to conduct PAIs 58% of the time in FY2020 Q3, and 64% of the time in FY2020 Q4.
- “MRA authority was used to assess 183 sites through MRA inspection reports for a total of 745 sites (18% of the FY2020 CDER Site Catalog). For comparison, in FY2019 1,258 drug quality assurance inspections were performed and an additional 109 sites were assessed using MRAs for a total of 1,367 sites (32% of the FY2020 CDER Site Catalog).”
3. The number of Warning Letters issued in FY2020 was slightly lower than in FY2018 or FY2019, but still over four times higher than in FY2015.
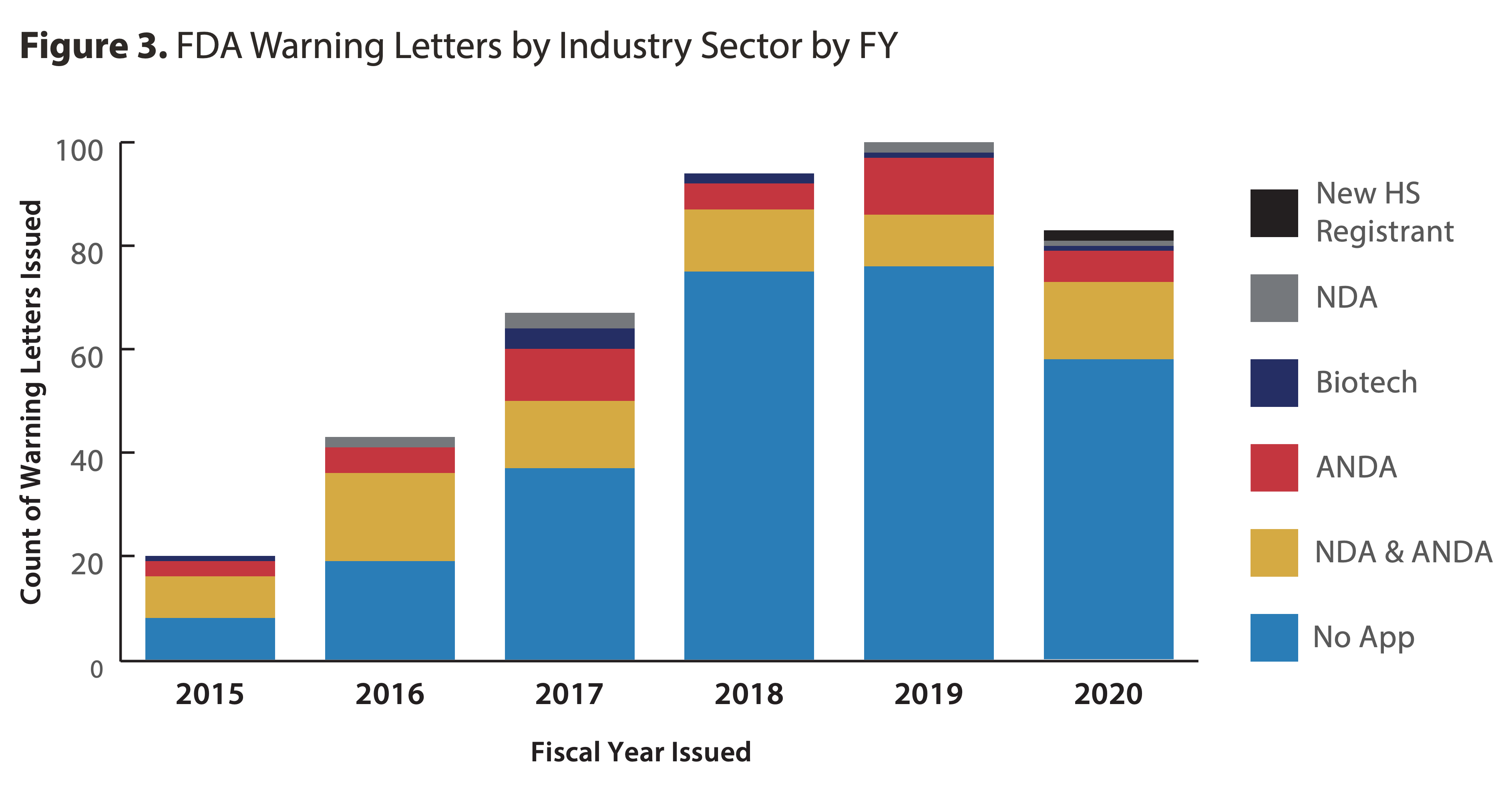
Source: fda.gov
- “As in past years, the majority of Warning Letters in FY2020 were issued to sites with non-application products (69%), and especially those that manufacture finished dosage form (FDF), non-sterile, non-application products (41% of all Warning Letters).”
- “Import Alerts doubled to 128 in FY2020. Latin America had the most sites on Import Alert for the first time in FY2020, due to an unprecedented number of new hand sanitizer registrants from Mexico that failed to meet quality standards."
4. Since FY2019, there has been a small decrease (0.10) in the mean Site Inspection Score (SIS) of the entire inventory of sites (7.3).
FDA’s SIS, a scale of 1 to 10, is used as a proxy for compliance with CGMP regulations. The SIS is based on the classification of FDA drug quality assurance inspections conducted over the prior ten years, including inspections classified under the MRA program, which allows some global regulators to recognize reports from their counterparts’ inspections.
- “Since FY2019, there was a small decrease (0.10) in the mean SIS of the entire inventory of sites (7.3).”
- “The mean SIS for sites for the U.S. (7.62), the EU (7.59), and Canada (7.56) remained higher than the global average.”
- “The mean SIS for sites in China (7.23), India (6.87), and Latin America (6.69) remained lower than the global average, though the scores indicate an acceptable level of compliance to CGMP on average.”
- “When considering sectors, sites making homeopathic products have the lowest mean SIS (6.77).”
- Based on an AI-powered regression model, “the number of application and non-application products being manufactured at a site were the top two most important features associated with higher or lower SIS, respectively. These types of analyses enable FDA to better target and apply surveillance resources.”
- “For FY2016–FY2020, three defect categories account for 60% of all defects reported: Product Quality Questioned, Device Issues, and Packaging Issues.”
5. In FY2020, the number of recall events increased for the second consecutive year.
- “The most recalled products by United States Pharmacopeia Therapeutic Category USPTC reflect the major quality issues over the past five years.”
- “The most substantial increases in the number of recalls by industry sector in FY2020 were in the No Application and NDA & ANDA (i.e., sites manufacturing for both application types) sectors.” (See full report for additional details.)
- “The average SIS of sites reporting at least one recall continues to be below the overall average SIS, highlighting the relationship between recalls and CGMP compliance.”
- “Each major recall over the last five fiscal years was associated with microbial or chemical contamination/impurities; a focus area for the industry to improve quality.”
6. CDER will continue to seek to minimize long-standing problems such as drug shortages due to quality issues through proactive efforts including the New Inspection Protocol Project (NIPP) and Quality Management Maturity.
- The NIPP “is aimed at using standardized electronic inspection protocols to collect data in a structured manner. The protocols promote consistent and comprehensive coverage of critical areas of drug manufacturing and provide structured, data-rich reports.
- “The protocols include questions related to quality culture observed in facilities.”
- “In the future, FDA will have the ability to better understand how certain variables (e.g., location of the establishment, type of establishment) affect quality. As more data are collected through NIPP, these types of insights can inform future inspections, identify policy/outreach opportunities, and influence application-related decision making.”
With regard to Quality Management Maturity, “FDA is moving toward a rating system that incentivizes drug manufacturers to invest in quality and achieve higher levels of quality management maturity. This concept was proposed in the multi-agency report for Congress Drug Shortages: Root Causes and Potential Solutions.”
Resources and Next Steps for FDA-Regulated Companies
Browse the selection of resources below for relevant guidance and insights.
- How to Prepare for Remote FDA Inspections/Evaluations
- 2020 FDA Warning Letter & Inspection Observation Trends [Updated March 2021]
- 7 Key Takeaways from FDA's Pandemic Inspections FAQ Guidance
- Remote Auditing Best Practices & Checklist for Regulatory Compliance
- 5 Items to Stock in Your FDA Inspection War Room
Need expert help identifying and remediating shortfalls within your Quality Management System (QMS) or build/augment your Quality Assurance team with top talent?
We help thousands of life science companies outsource their QA projects and fill specialized roles through convenient consulting projects, staff augmentation, and FTE recruitment.
Contact us today and get the conversation started.

FREE WHITE PAPER
The Life Science Quality Leadership Report& Benchmark: 2020
Compliance, Performance & Resourcing Emerge as Top Priorities
